|
|
|
|
|
|
|
|
|
|
|
|
|
| Code [GitHub] | Paper [arXiv] |
Determining protein structures is crucial yet computationally expensive. While models like AlphaFold2 predict structures using sequence data, they often ignore additional crystallographic information. To address this, we propose CrysFormer, a transformer-based model that predicts electron density maps directly from Patterson maps, incorporating partial structure data when available. Our method achieves state-of-the-art accuracy with reduced computational requirements.
Proteins, composed of amino acids, are essential to cellular functions, and their structure determines their role. Current methods for structure determination—such as X-ray crystallography or machine learning algorithms like AlphaFold2—have limitations. Incorporating additional data, such as crystallographic patterns, can improve accuracy. Here, we present CrysFormer, which utilizes transformers for protein electron density prediction. Our model shows significant improvements over convolution-based methods and sets the stage for solving more complex crystallographic problems.
The key highlights of our study include:
CrysFormer is the first transformer model tailored for predicting electron density maps from Patterson maps.
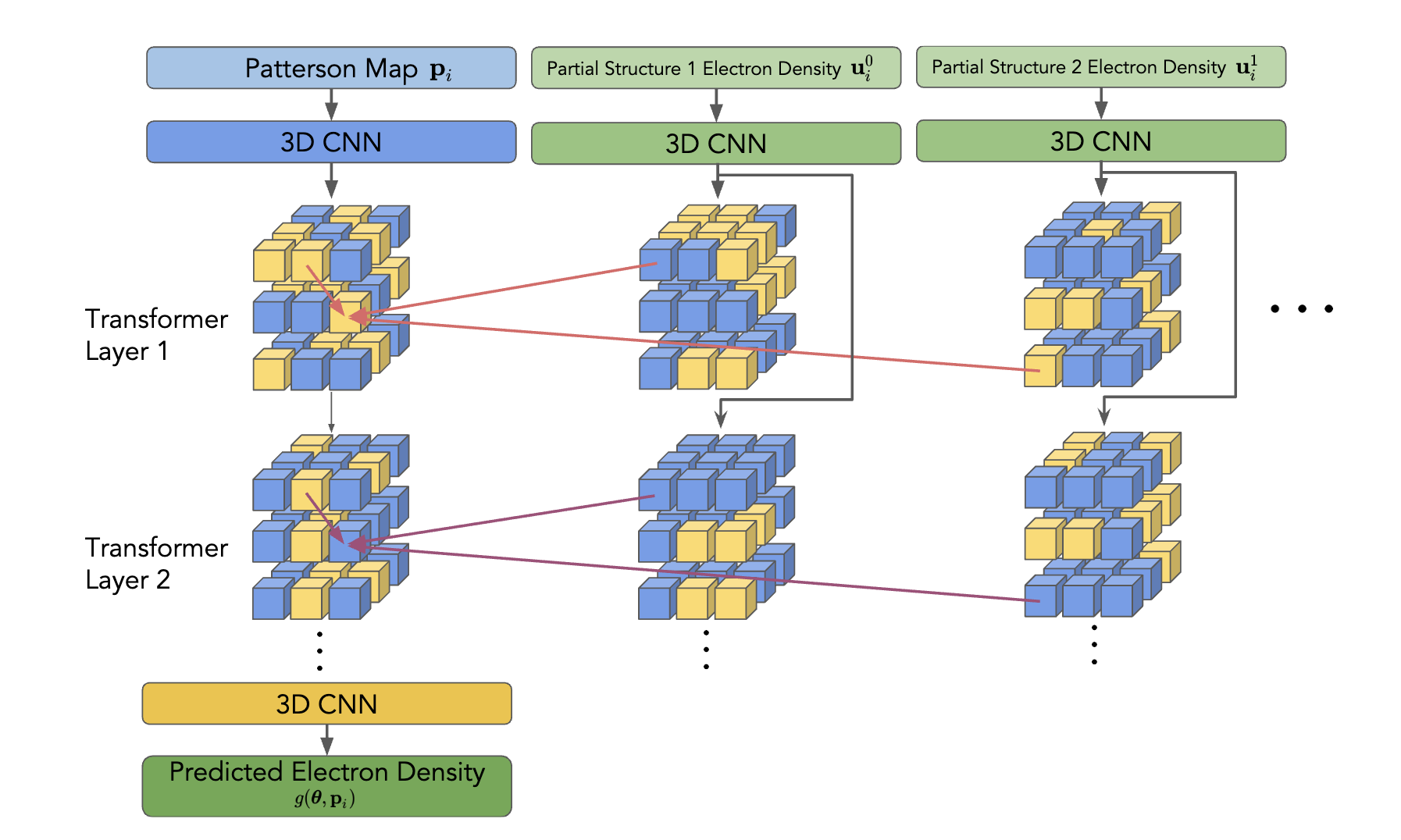
We leverage the Patterson function to preprocess crystallographic data and train a transformer-based model. Unlike convolutional models, CrysFormer employs an attention mechanism to capture global patterns in Patterson maps, integrating partial structure data for enhanced predictions. The model architecture includes efficient 3D patch embeddings and attention mechanisms optimized for 3D grids, ensuring scalability and accuracy.
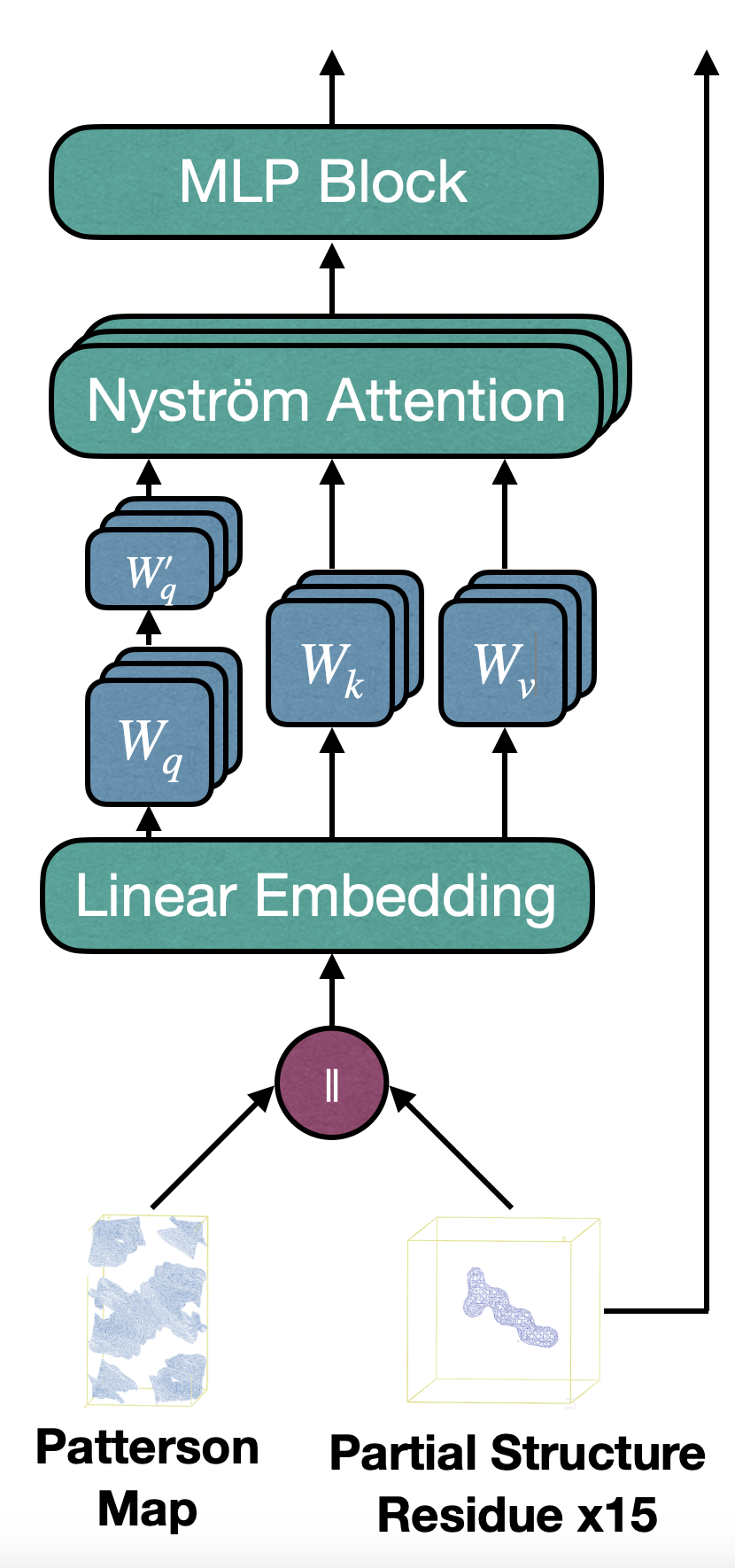
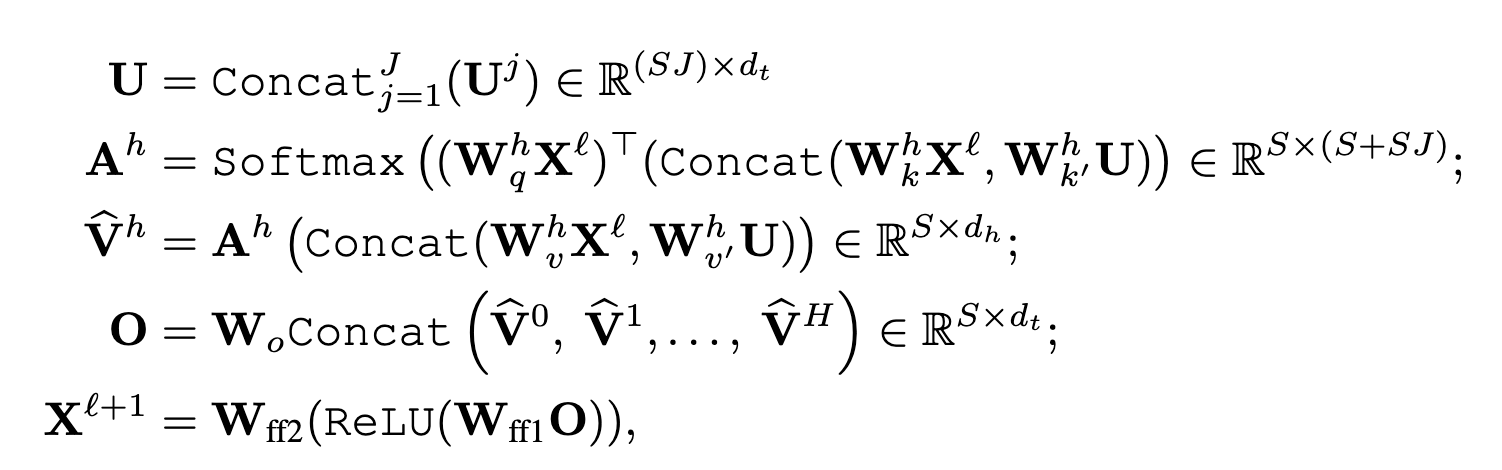
CrysFormer was benchmarked against enhanced convolutional networks on our datasets. Key findings include:
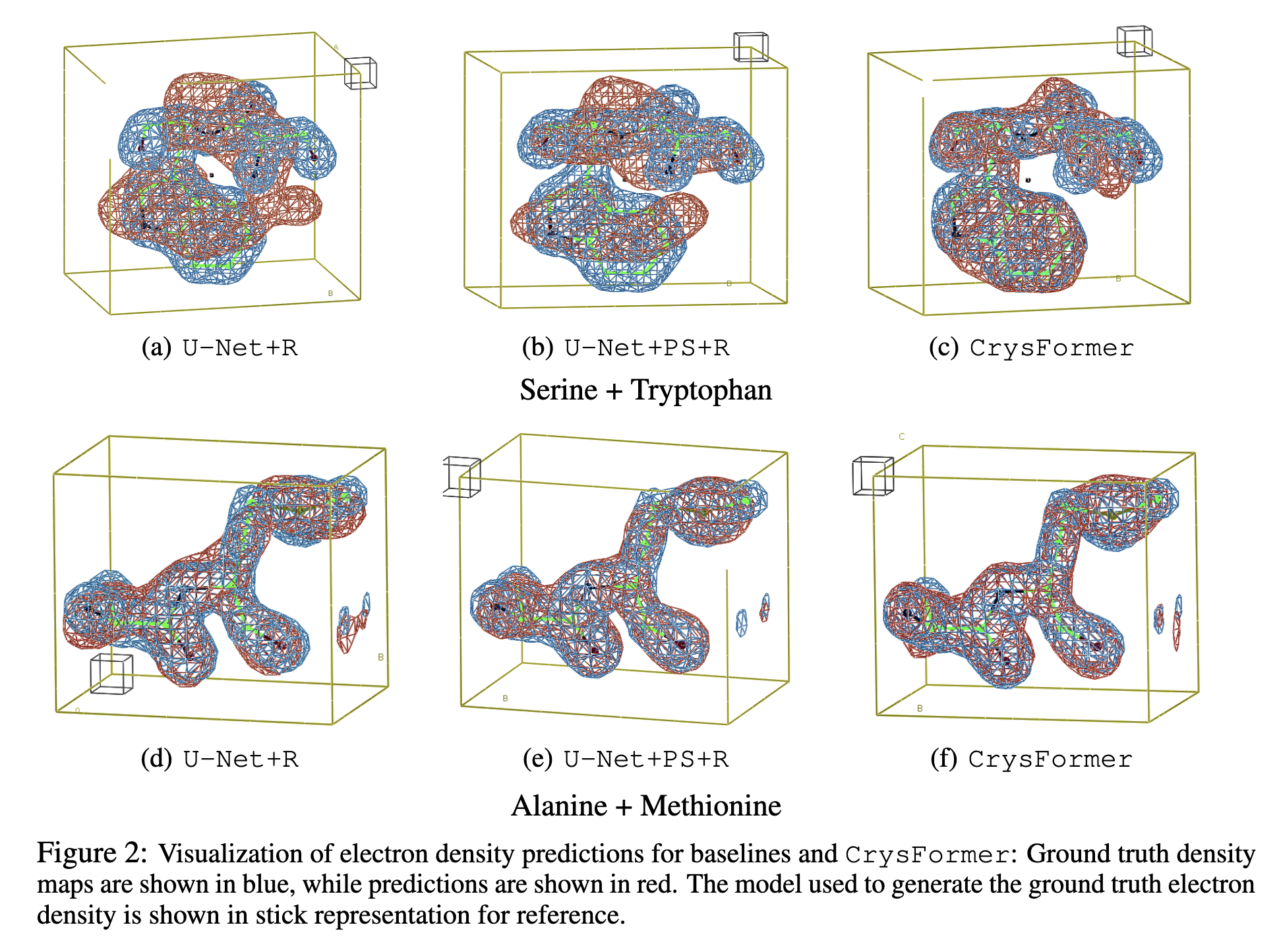
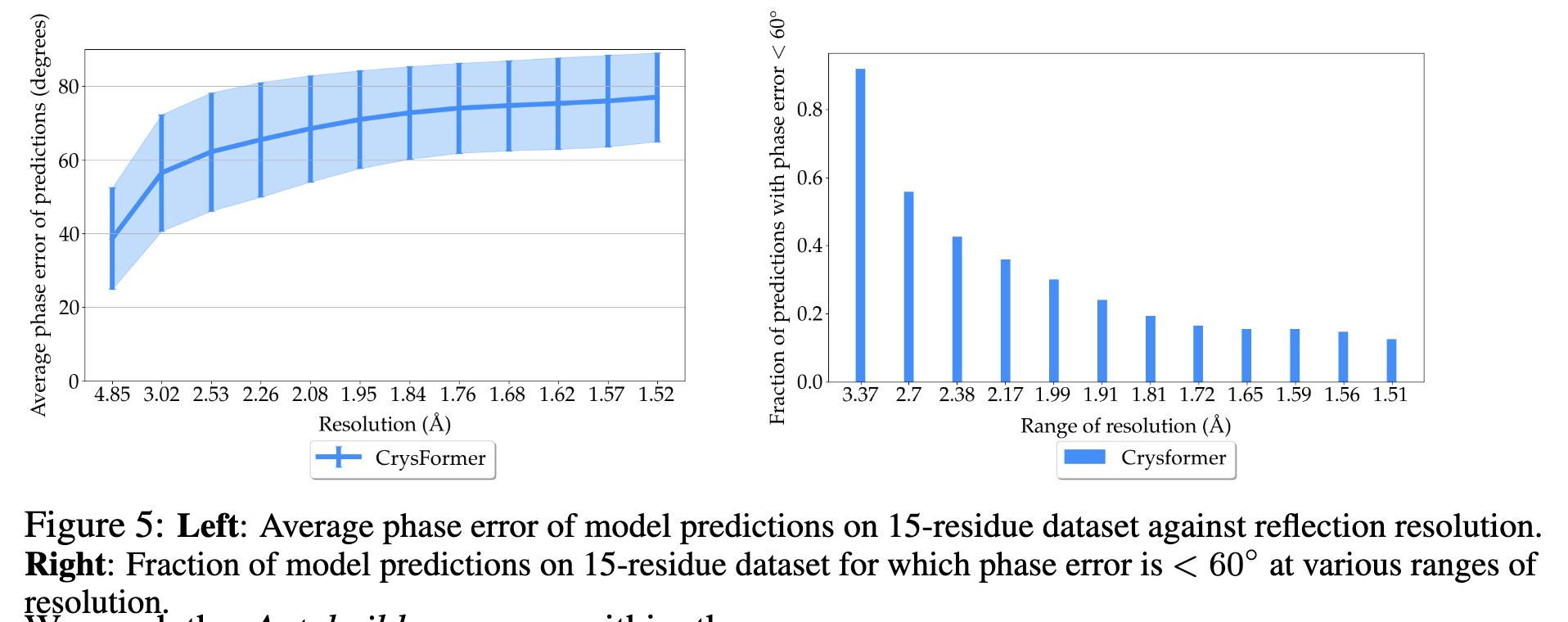
CrysFormer represents a significant advancement in protein crystallography, combining machine learning with domain-specific insights. Future work will explore variable unit cell geometries and extend the framework to larger protein structures. The method opens new avenues for computational biology, with potential applications in drug discovery and structural biology.
This research was supported by the Welch Foundation Grant A22-0307.