|
|
|
|
|
|
|
|
|
|
| Code [GitHub] | Paper [arXiv] |
Determining protein structures is crucial for understanding biological functions. Traditional methods like X-ray crystallography face challenges such as the phase problem. We present RecCrysFormer, a hybrid model that integrates transformers and convolutional layers to predict electron density maps directly from Patterson maps. Incorporating known partial structures and a recycling training regimen, our model achieves high accuracy on synthetic datasets, bridging experimental and computational approaches.
Proteins, composed of amino acids, play vital roles in biological processes. Understanding their 3D structure is essential for insights into their functionality. While experimental methods like X-ray crystallography are widely used, they face challenges, including the crystallographic phase problem. Machine learning approaches like AlphaFold2 have advanced the field but often neglect the use of raw experimental data like Patterson maps.
RecCrysFormer addresses these challenges by combining domain-specific knowledge with machine learning architectures. By leveraging Patterson maps, partial structures, and a novel recycling training regimen, it provides accurate electron density predictions, enabling seamless integration with established crystallographic refinement processes.
Our study introduces the following innovations:
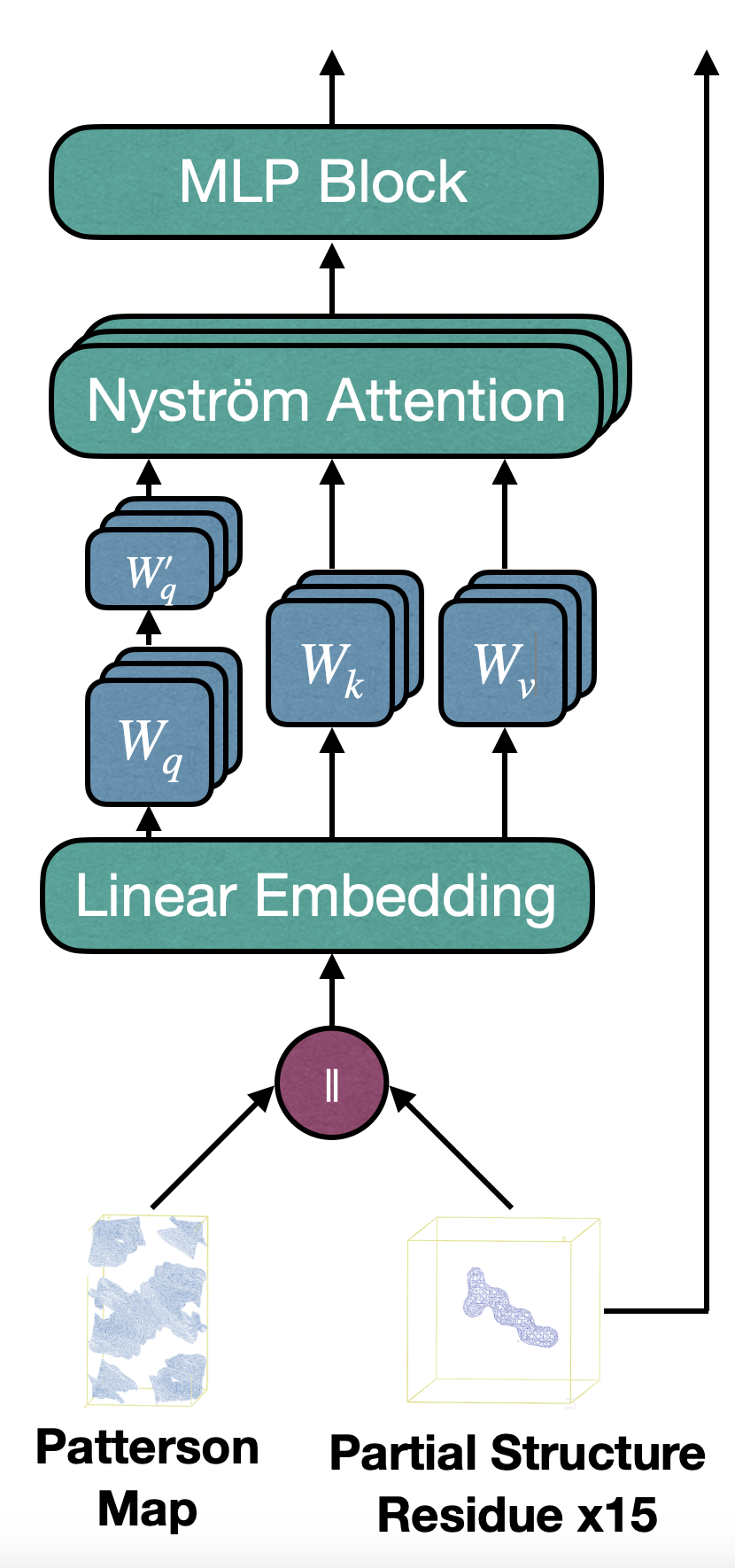
The core of RecCrysFormer is its ability to process raw experimental data in the form of Patterson maps, which capture interatomic vector relationships in protein crystals. These maps, while useful, lack direct information about atomic coordinates. To bridge this gap, RecCrysFormer employs a novel hybrid architecture combining 3D Convolutional Neural Networks (CNNs) and vision transformers.
The workflow begins with Patterson maps and, optionally, partial structure electron density maps as inputs. The Patterson maps are divided into small 3D patches, each transformed into feature tokens through convolutional layers and multi-layer perceptrons (MLPs). Positional embeddings are added to retain spatial information, and the resulting tokens are passed through a transformer core. The transformer uses attention mechanisms to capture global dependencies, allowing the model to integrate both Patterson maps and partial structures into coherent electron density predictions.
Key innovations include:
The loss function combines mean squared error (MSE) with the negative Pearson correlation coefficient, a crystallographic metric that captures phase accuracy and structural alignment. This hybrid loss promotes both general and domain-specific optimization, ensuring the model performs well on both global and detailed features.
RecCrysFormer also supports partial protein structures, which are derived from well-studied amino acid conformations. These are integrated as a secondary input, enabling the model to refine predictions based on known molecular patterns.
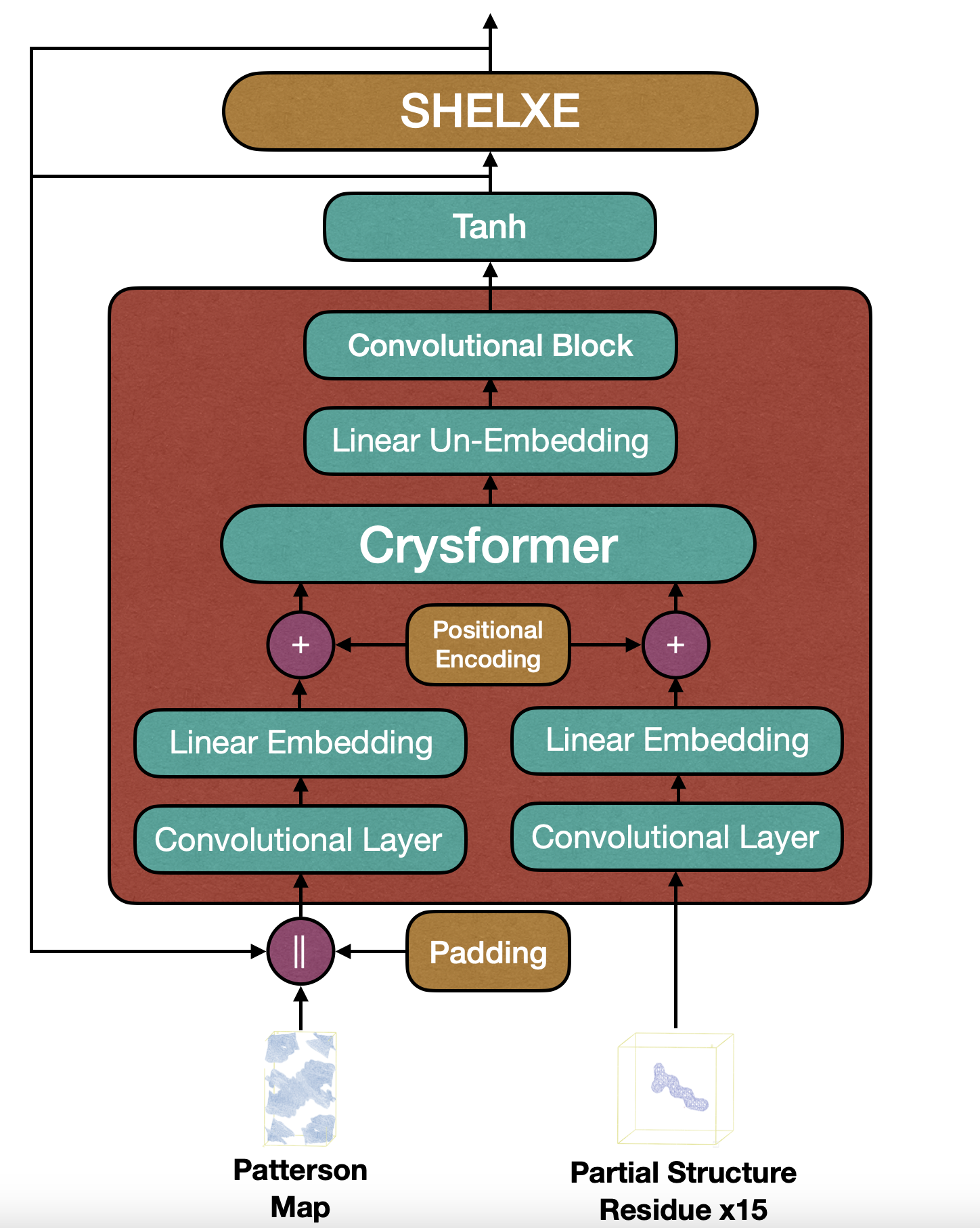
The final result is a powerful, data-efficient architecture capable of translating experimental Patterson maps into accurate electron density maps. This capability represents a significant step toward bridging the gap between experimental crystallographic methods and machine learning.
RecCrysFormer was benchmarked on synthetic datasets. Key findings include:
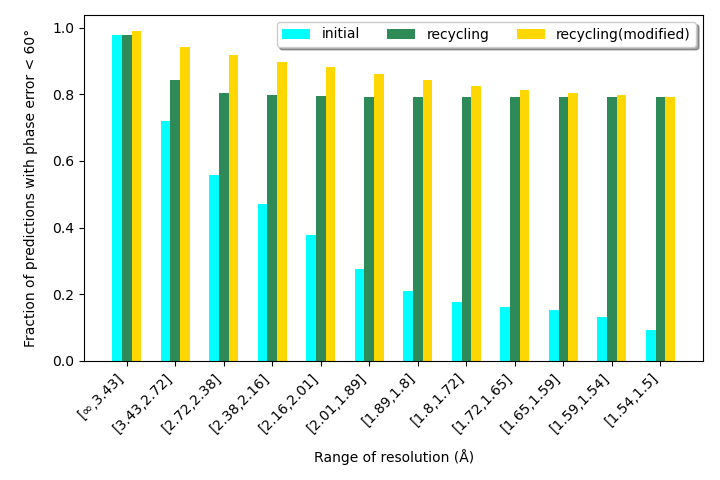
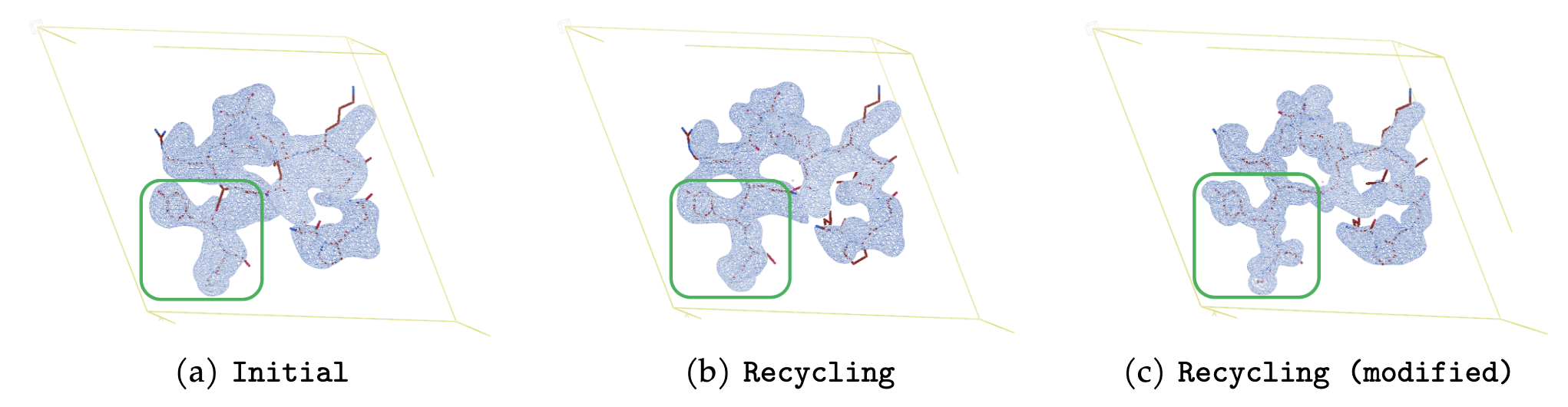

RecCrysFormer bridges experimental and computational methods, providing a robust framework for protein structure determination. Future work will involve scaling to full-length proteins, handling experimental noise, and extending the recycling procedure for enhanced performance.
This research was supported by the Welch Foundation Grant A22-0307 and the Ken Kennedy Institute at Rice University.